








La anemia microcítica cursa con hematíes de pequeño tamaño (microcitosis). Viene definida por la existencia de un volumen corpuscular medio (VCM) menor de 80 fL, aunque en niños y raza afroamericana puede ser menor. Generalmente se acompaña de hipocromía, definida por una hemoglobina corpuscular media (HCM) menor de 27 pg/célula. Puede existir microcitosis sin anemia.
2Etiología y clasificaciónExiste un descenso en la producción de hemoglobina que causa disminución en el tamaño de los hematíes y que puede ser secundario a: una alteración en la producción de las cadenas que forman la globina (talasemias), un déficit de hierro que dificulta su unión a la protoporfirina IX para formar el grupo hemo (anemia ferropénica), una alteración en la liberación del hierro existente que también impide la formación del grupo hemo (anemia de procesos inflamatorios) y otro tipo de defectos en la síntesis del grupo hemo1 (tabla 1).
Causas de anemia microcítica
| Anemia ferropénica2,3 | Por aumento de la demanda: en el embarazo, niños y adolescentes en crecimiento, atletasPor déficit de ingesta de hierro: dietas pobres en alimentos con hierroPor pérdidas de sangre:• Genitourinarias: menstruación abundante, metrorragias disfuncionales, sangrado por cáncer ginecológico, hematuria persistente por tumores vesicales• Digestivas: ulcus gastroduodenal, esofagitis por reflujo, ingesta de gastroerosivos (AINE, corticoides), angiodisplasia de colon, cáncer de colon, estómago, intestino delgado, varices esofágicas, enfermedad inflamatoria intestinal, parasitosis intestinales• Respiratorias: hemoptisis persistente en algunos procesos infecciosos o tumorales, hemosiderosis pulmonar idiopática, epistaxis de repetición• Politraumatizados, cirugía con sangrado abundante, donaciones o extracciones de sangre muy repetidasPor disminución de la absorción:• Enfermedad celiaca• Infección por Helicobacter pylori• Gastritis autoinmune• Gastrectomía, gastroyeyunostomía, cirugía bariátrica• Obesidad (por aumento de hepcidina)• Por fármacos: IBP, anti-H2 |
| Hemoglobinopatías hereditarias | α-talesemia:• α-talasemia silente, mínima o rasgo α-talasémico 1• α-talasemia minor o rasgo α-talasémico 2• α-talasemia intermedia o enfermedad de la hemoglobina H• α-talasemia major o síndrome de la hemoglobina de Bart con hidrops fetalis• α-talasemia y retraso mentalβ-talasemia:• β-talasemia minor o rasgo β-talasemia• β-talasemia intermedia•β-talasemia majorOtras β-hemoglobinopatías: hemoglobina E, hemoglobina E β-talasemia, hemoglobina Lepore, δ-β-talasemia, persistencia hereditaria de hemoglobina fetal |
| Trastornos genéticos del metabolismo del hierro4,5 | Anemia ferropénica refractaria a hierro (IRIDA): por defecto en TMPRSS6, produce déficit en la absorción del hierroMutaciones en DMT1 o SLC11A2Atransferrinemia: se altera el trasporte del hierroAceruloplasminemia: con diabetes mellitus, degeneración retiniana y enfermedad neurodegenerativa |
| Anemia de enfermedad o inflamación crónica | Procesos infecciosos crónicos: tuberculosis, endocarditis, osteomielitis, infección por VIHNeoplasiasEnfermedades autoinmunitarias: lupus eritematoso sistémico, artritis reumatoide, polimialgia reumática, vasculitis, sarcoidosisHipotiroidismo, insuficiencia renal crónica, hepatopatía crónica, EPOC, insuficiencia cardiaca, obesidad, enfermedad inflamatoria intestinal |
| Anemias sideroblásticas | Hereditarias o congénitas:• AS ligada al cromosoma X: aislada por mutación en ALAS2, AS ligada al cromosoma X con ataxia (por mutación en ABCB7)• AS con herencia autosómica recesiva: no sindrómicas por mutaciones en SLC25A38, GLRX5, HSPA9, y sindrómicas (AS con inmunodeficiencia de células B, fiebre periódica y retraso de desarrollo; AS con miopatía y acidosis láctica, AS sensible a tiamina con diabetes mellitus y sordera)• Formas heredadas mitocondriales: síndrome de Pearson de médula-páncreasAdquiridas:• Primarias: SMD con sideroblastos en anillo y displasia de linaje único, SMD con sideroblastos en anillo y displasia multilinaje, y SMD con sideroblastos en anillo y trombocitosis• Secundarias: por tóxicos (plomo, etanol), fármacos (isoniacida, pirazinamida, cloranfenicol, cicloserina, linezolid), o por déficit de piridoxina o cobre |
AINE: antiinflamatorios no esteroideos; Anti-H2: fármacos bloqueadores de los receptores H2; AS: anemia sideroblástica; EPOC: enfermedad pulmonar obstructiva crónica; IBP: inhibidores de la bomba de protones; SMD: síndrome mielodisplásico; VIH: virus de la inmunodeficiencia humana.
Fuente: elaboración propia.
Supone el 50% de las anemias6 y el 80% de casos de microcitosis. Es más frecuente en mujeres, sobre todo en edad fértil y embarazadas1. Puede existir ferropenia sin anemia.
Un adulto necesita 1-3mg de hierro al día, pero las mujeres en edad fértil, los niños y adolescentes en crecimiento y las embarazadas pueden necesitar hasta el doble. El hierro de los alimentos se absorbe en el intestino delgado. La hepcidina es importante en la regulación de la absorción del hierro y su posterior biodisponibilidad para la síntesis de hemoglobina. Niveles bajos de hepcidina (que aparecen en la ferropenia) favorecen la absorción intestinal del hierro y su movilización.
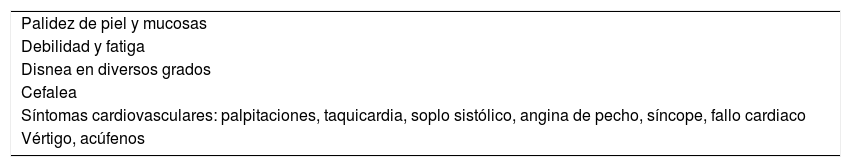
La anemia ferropénica (AF) suele presentar un curso crónico, por lo que muchas veces es asintomática o paucisintomática, lo que lleva a un infradiagnóstico. Los signos y síntomas son producidos tanto por la anemia en sí (tabla 2) como por la falta de hierro en tejidos y órganos (tabla 3).
Signos y síntomas específicos asociados a déficit de hierro
| Alteraciones cutáneas y mucosas |
| Alopecia difusa |
| Piel seca y áspera |
| Coiloniquia (uñas finas, frágiles, aplanadas o incluso cóncavas, en forma de cuchara) |
| Glositis atrófica |
| Queilitis angular |
| Alteraciones neurológicas y sensoriales |
| Síndrome de las piernas inquietas |
| Deterioro neurocognitivo |
| Pérdida auditiva |
| Pica: compulsión para ingerir sustancias que no son alimento, como hielo (pagofagia), tierra (geofagia), papel, tiza, almidón (amilofagia) |
| Beeturia: eliminación de orina roja o rosada tras la ingesta de remolacha (aparece hasta en un 14% de personas sanas y hasta en un 80% de personas con déficit de hierro) |
| Síndrome de Plummer-Vinson: cuadro de disfagia producido por la aparición de membranas o estenosis fibróticas a nivel faríngeo o esofágico alto en pacientes con anemia ferropénica evolucionada. Suele acompañarse de glositis, estomatitis angular, coiloniquia, y predispone al cáncer de esófago |
Fuente: elaboración propia.
Son trastornos hereditarios autosómicos recesivos que afectan a la síntesis de la hemoglobina al existir un déficit parcial o total en la producción de cadenas α o β de globina. Se estima que entre un 1% y un 5% de la población mundial es portadora de alguna mutación de talasemia11. Es más prevalente en zonas donde la malaria es o fue endémica (es un factor protector contra el paludismo), como son África subsahariana, cuenca mediterránea, Oriente Medio, subcontinente indio y sudeste asiático. Las formas más severas de α-talasemia se ven en el sudeste asiático y la cuenca mediterránea1. En España, la forma más frecuente es la β-talasemia minor.
La hemoglobina A (HbA) (más del 95% de la hemoglobina del adulto) está formada por 2 cadenas α y 2 cadenas β. El cromosoma 16 codifica la cade- na α mediante dos pares de alelos, mientras que el cromosoma 11 codifica la cadena β mediante un par de alelos. Diferentes tipos de deleciones o mutaciones pueden afectar a uno o más alelos, alterando de forma parcial o total la funcionalidad del alelo y produciendo un deterioro mayor o menor en la producción de la cadena de globina afectada. El desequilibrio en la proporción entre cadenas α y β llevará a la precipitación del exceso de cadenas no apareadas, dañándose las células precursoras de glóbulos rojos (sobre todo en la β-talasemia) y produciéndose eritropoyesis ineficaz. También se ven afectados por estas cadenas precipitadas los eritrocitos circulantes produciéndose hemólisis. Los individuos afectados presentarán diversos grados de anemia y hematopoyesis extramedular, que producirán cambios óseos, problemas de crecimiento y sobrecarga de hierro:
- •
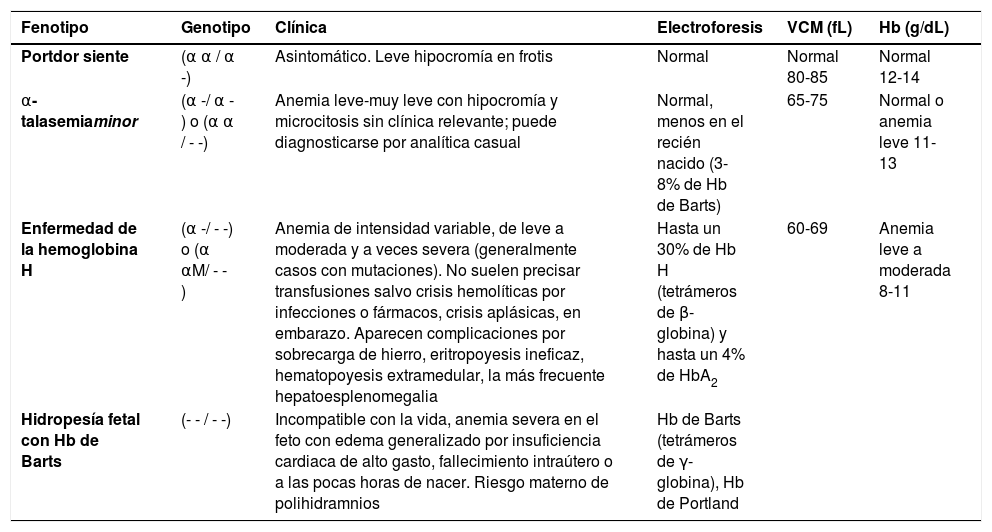
α-talasemia: existen más de 100 formas genéticas de α-talasemia con fenotipos que varían de asintomáticas a letales. Se pueden producir deleciones (-) y en menor medida mutaciones puntuales (αM) en uno, dos, tres o cuatro alelos, y suele correlacionarse bien el número de alelos afectados con la gravedad de la enfermedad12. Producen más gravedad clínica algunas mutaciones puntuales que afectan a la funcionalidad del alelo (como por ejemplo la Constant Spring) que la deleción de ese alelo13. La tabla 4 resume las características de los fenotipos de α-talasemia.
Tabla 4.Características de las α-talasemias1,12,13
Fenotipo Genotipo Clínica Electroforesis VCM (fL) Hb (g/dL) Portdor siente (α α / α -) Asintomático. Leve hipocromía en frotis Normal Normal 80-85 Normal 12-14 α-talasemiaminor (α -/ α -) o (α α / - -) Anemia leve-muy leve con hipocromía y microcitosis sin clínica relevante; puede diagnosticarse por analítica casual Normal, menos en el recién nacido (3-8% de Hb de Barts) 65-75 Normal o anemia leve 11-13 Enfermedad de la hemoglobina H (α -/ - -) o (α αM/ - -) Anemia de intensidad variable, de leve a moderada y a veces severa (generalmente casos con mutaciones). No suelen precisar transfusiones salvo crisis hemolíticas por infecciones o fármacos, crisis aplásicas, en embarazo. Aparecen complicaciones por sobrecarga de hierro, eritropoyesis ineficaz, hematopoyesis extramedular, la más frecuente hepatoesplenomegalia Hasta un 30% de Hb H (tetrámeros de β-globina) y hasta un 4% de HbA2 60-69 Anemia leve a moderada 8-11 Hidropesía fetal con Hb de Barts (- - / - -) Incompatible con la vida, anemia severa en el feto con edema generalizado por insuficiencia cardiaca de alto gasto, fallecimiento intraútero o a las pocas horas de nacer. Riesgo materno de polihidramnios Hb de Barts (tetrámeros de γ-globina), Hb de Portland Hb: hemoglobina; VCM: volumen corpuscular medio.
Fuente: elaboración propia.
- •
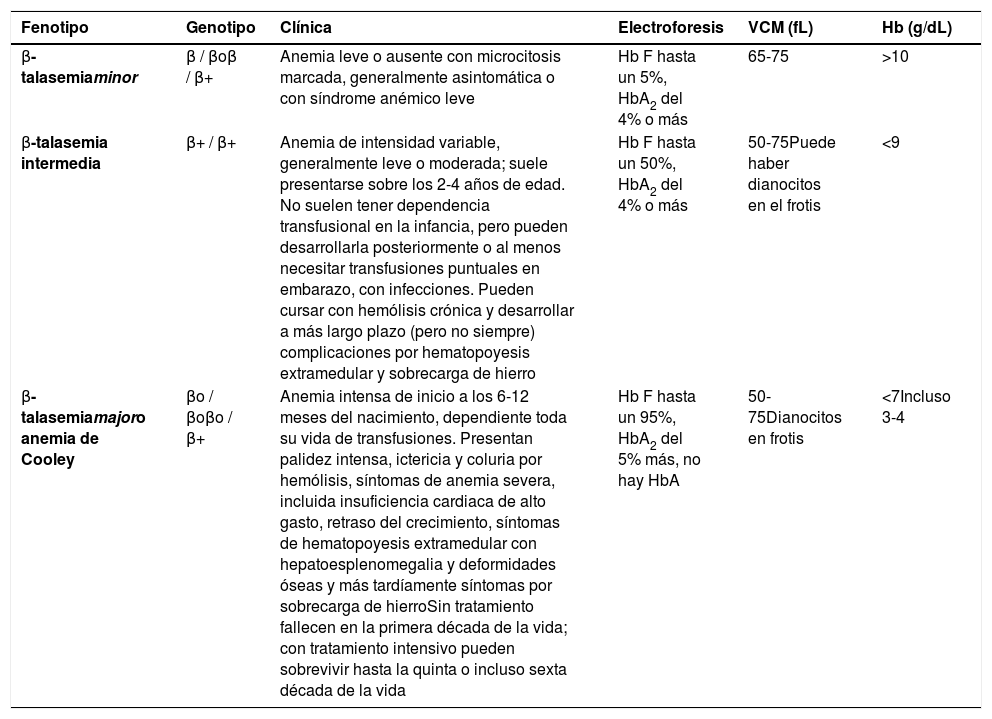
β-talasemia: se produce como consecuencia de mutaciones en uno o ambos alelos, mutaciones que pueden ser silentes (β silentes), reducir la producción de globina β (β+) o eliminar totalmente su producción (βo)11. La gravedad se correlaciona con la alteración en la síntesis de cadenas β. Los síntomas no aparecen en el recién nacido, sino a partir de los 4-6 meses, cuando las cadenas γ de la hemoglobina fetal se sustituyen por las cadenas β de la HbA. Las características de los tipos de β-talasemia se resumen en la tabla 5.
Tabla 5.Características de las β-talasemias1,13
Fenotipo Genotipo Clínica Electroforesis VCM (fL) Hb (g/dL) β-talasemiaminor β / βoβ / β+ Anemia leve o ausente con microcitosis marcada, generalmente asintomática o con síndrome anémico leve Hb F hasta un 5%, HbA2 del 4% o más 65-75 >10 β-talasemia intermedia β+ / β+ Anemia de intensidad variable, generalmente leve o moderada; suele presentarse sobre los 2-4 años de edad. No suelen tener dependencia transfusional en la infancia, pero pueden desarrollarla posteriormente o al menos necesitar transfusiones puntuales en embarazo, con infecciones. Pueden cursar con hemólisis crónica y desarrollar a más largo plazo (pero no siempre) complicaciones por hematopoyesis extramedular y sobrecarga de hierro Hb F hasta un 50%, HbA2 del 4% o más 50-75Puede haber dianocitos en el frotis <9 β-talasemiamajoro anemia de Cooley βo / βoβo / β+ Anemia intensa de inicio a los 6-12 meses del nacimiento, dependiente toda su vida de transfusiones. Presentan palidez intensa, ictericia y coluria por hemólisis, síntomas de anemia severa, incluida insuficiencia cardiaca de alto gasto, retraso del crecimiento, síntomas de hematopoyesis extramedular con hepatoesplenomegalia y deformidades óseas y más tardíamente síntomas por sobrecarga de hierroSin tratamiento fallecen en la primera década de la vida; con tratamiento intensivo pueden sobrevivir hasta la quinta o incluso sexta década de la vida Hb F hasta un 95%, HbA2 del 5% más, no hay HbA 50-75Dianocitos en frotis <7Incluso 3-4 Hb: hemoglobina; VCM: volumen corpuscular medio.
Fuente: elaboración propia.
- •
Actualmente se habla de talasemias dependientes de transfusiones (TDT) (requieren transfusiones de forma continua para sobrevivir) que incluyen la β-talasemia mayor y formas severas y evolucionadas de β-talasemia intermedia y de enfermedad de la hemoglobina H, y talasemias no dependientes de transfusiones (TNDT) (pueden precisar puntualmente transfusiones en situaciones especiales) que incluyen la mayoría de los casos de β-talasemia intermedia y de enfermedad de la hemoglobina H11. La hemólisis, la eritropoyesis ineficaz y subsecuente hematopoyesis extramedular, y la sobrecarga de hierro tanto primaria (por aumento de absorción intestinal) como secundaria a transfusiones, van a producir diversas complicaciones (tabla 6).
Tabla 6.Manifestaciones clínicas de las talasemias11,13
Secundarias a hemólisis IctericiaCrisis hemolíticas agudasColelitiasis Cambios esqueléticos (por expansión medular y hematopoyesis extramedular) Trastornos del crecimiento: acortamiento de extremidades, ensanchamiento óseoDeformidad facialDolor óseoOsteoporosisSeudotumores óseos. Pueden producir compresión de nervios, médula espinal Hepatoesplenomegalia Alteraciones secundarias a depósito de hierro en diversos órganos Fibrosis hepática, cirrosis hepática, cáncer de hígadoEndocrinopatías: hipogonadismo (por depósito de hierro en hipófisis), hipotiroidismo, insulinorresistencia y diabetes, alteración del crecimientoFallo cardiaco, arritmias, secundario a miocardiopatía por depósito de hierro sumado a la anemia, la hipoxia tisular y la hipertensión pulmonar Hiperuricemia Hipercoagulabilidad Trombosis venosa profundaTromboembolismo pulmonarIctus Hipertensión pulmonar Úlceras en piernas Mayor incidencia de infecciones Hepatitis víricas, infección por VIHMayor predisposición a infecciones sobre todo en esplenectomizados VIH: virus de la inmunodeficiencia humana.
Fuente: elaboración propia.
Es la segunda causa de anemia. Aunque suele cursar como una anemia normocítica, hasta en una cuarta parte de los pacientes puede ser microcítica. La inflamación persistente favorece la liberación de citoquinas que causan una disminución relativa en la producción de eritropoyetina y una disminución en la capacidad de respuesta de la médula ósea a la misma. Además, existe un aumento de hepcidina (reactante de fase aguda) que favorece la microcitosis1,14.
Es una anemia hiporregenerativa, de intensidad leve a moderada, generalmente oligosintomática, en la que predominan los síntomas de la enfermedad de base.
2.4Anemia sideroblástica15Anemia poco frecuente que incluye procesos tanto hereditarios como adquiridos en los que se produce una mala utilización del hierro en la eritropoyesis por diversas alteraciones en la síntesis del hemo y en la función mitocondrial:
- •
Las anemias sideroblásticas (AS) congénitas o hereditarias pueden manifestarse de forma aislada (lo más frecuente) o dentro de un síndrome con diferentes manifestaciones clínicas. Los defectos genéticos pueden producir alteraciones en tres vías metabólicas mitocondriales: la síntesis del hemo, la producción de grupos hierro-azufre y la síntesis de algunas proteínas. Suelen diagnosticarse en la infancia o la adolescencia. Los niños con enfermedad grave pueden tener problemas de desarrollo y crecimiento. La AS ligada a X por mutaciones heterocigotas en ALAS2 es la más frecuente. Afecta más a varones que a mujeres, con una anemia de severidad variable. En varones es microcítica, pero en mujeres es normocítica o macrocítica. Las AS con herencia autosómica recesiva suelen ser microcíticas (salvo la sensible a tiamina que es megaloblástica) y cursan con anemia moderada-severa (más leve en mutaciones de HSPA9). Las formas heredadas mitocondriales cursan con anemia severa normo o macrocítica.
- •
Las AS adquiridas pueden deberse a una alteración primaria en las células madre de la médula ósea (AS clonales) o ser secundarias (tabla 1).
- –
Las AS clonales se consideran variantes de síndromes mielodisplásicos y neoplasias mieloproliferativas. Son las más frecuentes y suelen afectar a mayores de 40 años. Producen una anemia moderada normo o macrocítica de curso insidioso. Pueden descubrirse de forma incidental o al estudiar a pacientes con anemia, descompensación de su patología cardiaca, alteraciones en la serie blanca y plaquetaria (variantes con displasia multilinaje y con trombocitosis) o con hepatoesplenomegalia. La mutación adquirida más frecuente en estos pacientes es en SF3B1.
- –
Las AS secundarias son reversibles cuando se elimina la causa. Suelen ser normo-macrocíticas salvo la producida por isoniacida. El abuso de alcohol produce anemia multifactorial y hasta en un tercio encontraremos cambios sideroblásticos. El déficit de cobre puede ser provocado por malabsorción, dieta parenteral prolongada o enteral sin suplementos de cobre, uso de quelantes del cobre como la trientina, o consumo excesivo de zinc. Produce una anemia progresiva y a veces muy severa, con eritrocitos de diversos tamaños, reversible tras tratamiento y asociada a síntomas neurológicos menos reversibles.
- –
La intoxicación por plomo produce anemia que algunos autores incluyen en AS secundarias.
- –
Raro trastorno genético autosómico recesivo por mutaciones en el gen TMPRSS6 del cromosoma 22, que produce un defecto en la inhibición de producción de hepcidina. Cursa con niveles inapropiadamente altos de hepcidina en relación con la hiposideremia que sufren. Tienen su inicio en la infancia (2-4 años) con una anemia moderada (6-9g/dL), con importante microcitosis (45-65 fL), saturación de transferrina muy baja, y falta de respuesta al tratamiento con hierro oral. Responden al tratamiento con hierro parenteral, aunque no de forma completa. No presentan habitualmente alteraciones del desarrollo físico ni intelectual y manifiestan pocos estigmas de ferropenia. Suele disminuir la severidad de la anemia en la adolescencia y edad adulta.
3DiagnósticoAnte un paciente con anemia microcítica deberemos realizar una anamnesis y exploración adecuada que incluya edad, etnia, orígenes geográficos, antecedentes familiares de anemia y enfermedades hereditarias, ingesta de fármacos, alcohol y otros tóxicos, encuesta nutricional, síntomas digestivos, posibles sangrados de diversos orígenes, posibles patologías crónicas y neoplasias concurrentes, sintomatología sistémica (fiebre, pérdida de peso, sudoración nocturna), y síntomas y signos específicos (pica, queilitis angular, coiloniquia, organomegalias, presencia de estigmas hepáticos)17,18.
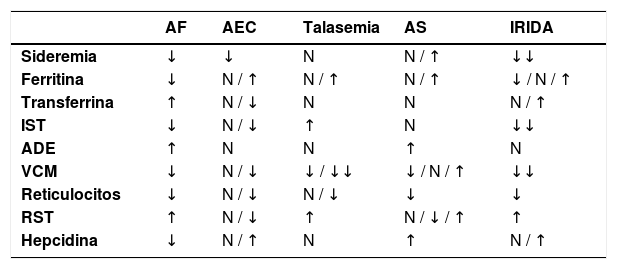
Solicitaremos un hemograma completo incluyendo recuento de eritrocitos, hemoglobina, VCM, HCM, CHCM, ADE (amplitud de distribución eritrocitaria) y número de reticulocitos. Solicitaremos sideremia (parámetro de poco valor), niveles de transferrina (normal 200-400mg/dL), capacidad total de fijación de hierro (CTFH), índice de saturación de transferrina (IST) (normal 20-50%) y ferritina sérica. Esta última es la que mejor refleja la reserva de hierro existente, de forma que niveles de ferritina por debajo de 15-30 ng/mL son indicativos de déficit de hierro en personas sanas, pero al ser un reactante de fase aguda es probable que personas con enfermedad hepática, neoplasias y procesos infecciosos o inflamatorios puedan tener ferropenia con cifras de ferritina de 50 o incluso 100 ng/mL17–19. Otro parámetro útil para distinguir AF de AEC es el receptor soluble de la transferrina (RST) (no se afecta por la inflamación). La ratio entre RST y el logaritmo de la ferritina discrimina bien si existe déficit de hierro: valores por debajo de 1 sugerirían AEC y valores por encima de 2, AF. La tabla 7 refleja la situación de estos parámetros en las diferentes causas de anemia microcítica.
Parámetros analíticos según la causa de la anemia microcítica
| AF | AEC | Talasemia | AS | IRIDA | |
|---|---|---|---|---|---|
| Sideremia | ↓ | ↓ | N | N / ↑ | ↓↓ |
| Ferritina | ↓ | N / ↑ | N / ↑ | N / ↑ | ↓ / N / ↑ |
| Transferrina | ↑ | N / ↓ | N | N | N / ↑ |
| IST | ↓ | N / ↓ | ↑ | N | ↓↓ |
| ADE | ↑ | N | N | ↑ | N |
| VCM | ↓ | N / ↓ | ↓ / ↓↓ | ↓ / N / ↑ | ↓↓ |
| Reticulocitos | ↓ | N / ↓ | N / ↓ | ↓ | ↓ |
| RST | ↑ | N / ↓ | ↑ | N / ↓ / ↑ | ↑ |
| Hepcidina | ↓ | N / ↑ | N | ↑ | N / ↑ |
ADE: amplitud de distribución eritrocitaria; AEC: anemia de enfermedad crónica; AF: anemia ferropénica; AS: anemia sideroblástica; IRIDA: anemia ferropénica resistente a hierro; IST: índice de saturación de transferrina; RST: receptor soluble de la transferrina; VCM: volumen corpuscular medio.
Modificado de Sebastián E y Sevilla J8.
En el frotis de sangre periférica algunos hallazgos orientan sobre la etiología de la microcitosis. En la AF encontraremos anisocitosis e hipocromía, presencia de ovalocitos (células en lápiz), poiquilocitos, eliptocitos y dacriocitos. En la talasemia hay menos anisocitosis, pueden aparecer también ovalocitos y poiquilocitos, punteado basófilo y en formas severas dacriocitos y dianocitos (bastante característicos). En AS encontraremos anisocitosis, poiquilocitosis, algún dianocito y las células más carasterísticas son los siderocitos (eritrocitos hipocrómicos con cuerpos de Pappenheimer). En la anemia por intoxicación por plomo es típico el punteado basófilo.
La tabla 8 presenta el enfoque diagnóstico de la anemia microcítica. En algunos casos pueden coincidir varias causas de anemia, apareciendo en las pruebas resultados no esperados.
Enfoque diagnóstico de la anemia microcítica
| Presencia de ferropenia | ADE alto | Si ferritina baja, IST bajo o responde a hierro oral: anemia ferropénicaEstudiar etiología, sobre todo en varones y mujeres posmenopáusicas | |
| Si no responde a hierro oral | Si existe malabsorción: anemia ferropénica | ||
| Si no existe malabsorción, es niño, con IST muy bajo, ferritina normal o baja, ratio hepcidina:IST alta, sospechamos IRIDAEl análisis del gen TMPRSS6 confirma el diagnóstico4 | |||
| ADE normal | Si niveles de ferritina normales o altos no explicables por inflamación aguda (ratio RST-logaritmo de ferritina normal)Proceso crónico/inflamatorio de base: PCR y VSG generalmente elevadasSospecha de anemia de enfermedad crónica | ||
| Ausencia de ferropenia | ADE alto | Si descartamos causas frecuentes de microcitosis, frotis sugestivo, realizar análisis de médula ósea con tinción de Perls: si ≥ 15% de sideroblastos en anillo, confirmamos anemia sideroblásticaSi es menor 20 años, probable AS congénita: pedir pruebas genéticas y valorar sintomatología sindrómicaSi es adulto generalmente con AS adquirida: suelen ser normo-macrocíticas salvo si es por isoniacida que puede ser microcítica4 | |
| ADE normal | Si VCM muy bajo respecto al grado de anemia (con número de eritrocitos incluso normal o alto): sospechar talasemiaSolicitar análisis de hemoglobinas mediante electroforesis o técnicas de cromatografía liquida de alta resolución (tablas 4 y 5)Si α-talasemia (sobre todo minor o mínima), el análisis genético de ADN es la única prueba válida para diagnósticoLa confirmación diagnóstica la dará la prueba del gen de la globina11–13 |
ADE: amplitud de distribución eritrocitaria; ADN: ácido desoxirribonucleico; AS: anemia sideroblástica; IRIDA: anemia ferropénica refractaria a hierro; IST: índice de saturación de transferrina; PCR: proteína C reactiva; RST: receptor soluble de transferrina; VCM: volumen corpuscular medio; VSG: velocidad de sedimentación globular.
Fuente: elaboración propia.
Además de tratar la causa que produce el déficit de hierro, deberemos dar suplementos de hierro: la vía oral es la de elección (por precio y efectividad) en la mayoría de los casos. Solo en pacientes con anemia severa y síntomas cardiovasculares estará indicada la transfusión de concentrados de hematíes, que además de corregir la hipoxia aportan 200mg de hierro por cada unidad3.
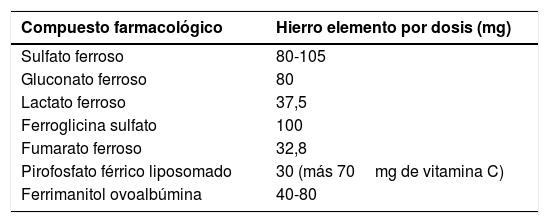
Hierro oral: administrar sales ferrosas que tienen mejor biodisponibilidad; es de elección el sulfato ferroso, del que existen diversos preparados (tabla 9). Se recomiendan dosis de 100-200mg (3-6mg/kg de peso en niños) de hierro elemental, con el estómago vacío, mejor con vitamina C que mejora la absorción, en una o dos dosis, manteniendo el tratamiento 3-6 meses para conseguir la repleción de depósitos de hierro3. Existe la posibilidad de administrar las dosis cada 48 horas para mejorar tolerancia y absorción7. Realizar control de hemoglobina a las 4 semanas, y si no se eleva al menos 1g/dL se considerará fallo de tratamiento, que puede ser debido a resistencia al tratamiento (por malabsorción, IRIDA), falta de adherencia o mala tolerancia (los efectos secundarios más frecuentes son náuseas y vómitos, diarrea o estreñimiento, molestia epigástrica, sabor metálico y heces negras).
Formulaciones de hierro oral7,20
| Compuesto farmacológico | Hierro elemento por dosis (mg) |
|---|---|
| Sulfato ferroso | 80-105 |
| Gluconato ferroso | 80 |
| Lactato ferroso | 37,5 |
| Ferroglicina sulfato | 100 |
| Fumarato ferroso | 32,8 |
| Pirofosfato férrico liposomado | 30 (más 70mg de vitamina C) |
| Ferrimanitol ovoalbúmina | 40-80 |
Fuente: elaboración propia.
Hierro intravenoso: la eficacia y rapidez de acción, la mayor seguridad de los nuevos preparados y su comodidad posológica (una dosis única muchas veces) hacen de la terapia intravenosa el tratamiento de elección en diversas situaciones (tabla 10). La dosis a administrar se calcula por la fórmula da Garzoni:
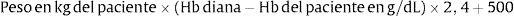
Indicaciones del tratamiento con hierro intravenoso3,7,20
| Indicaciones establecidas |
| Fracaso del tratamiento oral: por intolerancia al hierro oral, situaciones con malabsorción, mal cumplimiento, IRIDA |
| Enfermedad inflamatoria intestinal en fase activa |
| Anemia severa con Hb < 8 g/dL |
| Pacientes con necesidad de rápida recuperación: pacientes con AF y fallo cardiaco, embarazadas en 2.° y 3.er trimestre con anemia severa, pacientes con sangrado crónico que no remontan cifras de hemoglobina con hierro oral |
| Anemia en insuficiencia renal crónica en tratamiento con análogos de eritropoyetina |
| Pacientes con indicación de transfusión sanguínea que la rechazan por motivos religiosos |
| Indicaciones potenciales |
| Ancianos con AF |
| Anemia en insuficiencia renal crónica sin tratamiento con análogos de eritropoyetina |
| Pacientes con fallo cardiaco y ferropenia sin anemia significativa |
| Programas de ahorro de sangre en pacientes quirúrgicos, anemia perioperatoria |
| Pacientes con anemia y cáncer, quimioterapia, enfermedades crónicas, que no responden adecuadamente solo a tratamiento con análogos de eritropoyetina |
| Sangrado uterino severo |
AF: anemia ferropénica; Hb: hemoglobina; IRIDA: anemia ferropénica refractaria a hierro.
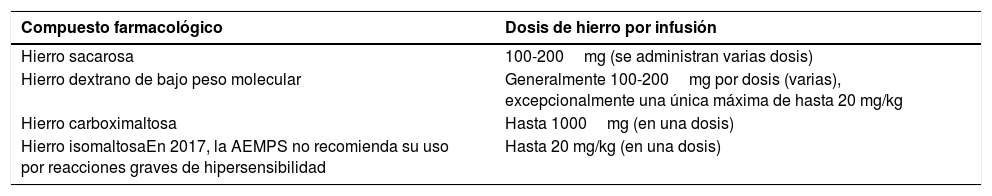
Normalmente no pasa de 1000-1500mg. Los efectos secundarios más frecuentes son náuseas y vómitos, cefalea, prurito y flushing, artromialgias y dolor torácico, y suelen ser transitorios. Actualmente son raras las reacciones severas de tipo anafiláctico. Los preparados utilizados se reflejan en la tabla 11.
Preparados de hierro intravenoso disponibles en España20
| Compuesto farmacológico | Dosis de hierro por infusión |
|---|---|
| Hierro sacarosa | 100-200mg (se administran varias dosis) |
| Hierro dextrano de bajo peso molecular | Generalmente 100-200mg por dosis (varias), excepcionalmente una única máxima de hasta 20 mg/kg |
| Hierro carboximaltosa | Hasta 1000mg (en una dosis) |
| Hierro isomaltosaEn 2017, la AEMPS no recomienda su uso por reacciones graves de hipersensibilidad | Hasta 20 mg/kg (en una dosis) |
AEMPS: Agencia Española de Medicamentos y Productos Sanitarios.
Modificado de De la Iglesia20.
Los objetivos son corregir la anemia, reducir la hematopoyesis extramedular y la expansión medular, prevenir o tratar la sobrecarga de hierro y tratar las complicaciones. Los pacientes con talasemia minor o mínima no requieren tratamiento. En la tabla 12 se exponen los tratamientos para talasemia intermedia y major.
Opciones terapéuticas en talasemia
| Transfusión11,21 |
| De forma programada, regular, y durante toda la vida, desde la infancia en β-talasemia mayor (TDT) y cuando se considere en casos de talasemia intermedia (el paso de TNDT a TDT puede ser transitorio o definitivo) |
| De forma ocasional de TNDT: infecciones agudas, embarazo, cirugía |
| Mantener, con el régimen de transfusión programada, hemoglobina por encima de 9-10,5 g/dL (11-12 g/dL en cardiópatas), llegando a 12-13 g/dL (nunca más de 15) tras transfusión |
| Generalmente se pasará de TNDT a TDT si el paciente presenta compromiso cardiopulmonar, deterioro funcional o síntomas significativos de hematopoyesis extramedular. No suele ser necesario si el paciente mantiene hemoglobinas por encima de 9-10 g/dL |
| Efectos secundarios: sobrecarga de hierro, aloinmunización, transmisión de infecciones, reacciones alérgicas, reacciones febriles no hemolíticas, sobrecarga circulatoria, lesión pulmonar aguda |
| Quelación del hierro11,21 |
| En talasemia mayor se usa ya en la infancia, desde el inicio del programa de transfusiones regulares; en la intermedia puede ser necesaria a partir de la 2.ª o 3.ª década de vida |
| Vigilar la sobrecarga de hierro midiendo la ferritina sérica periódicamente (debe mantenerse por debajo de 1000 ng/mL), y medir la carga de hierro en hígado y miocardio por técnicas de resonancia magnética |
| Se utilizan deferoxamina parenteral y deferiprona o deferasirox por vía oral |
| Esplenectomía11,21 |
| Actualmente, uso restringido a: pacientes que no pueden recibir transfusiones o quelantes del hierro, esplenomegalia sintomática, hiperesplenismo que produce citopenias graves, infarto esplénico o trombosis de vena esplénica, anemia severa que no se controla con régimen intensivo de transfusiones |
| Efectos secundarios importantes: riesgo aumentado de infecciones y sepsis (realizar vacunaciones y tratamiento antibiótico profiláctico) y mayor riesgo tromboembólico (profilaxis con AAS o incluso HBPM) |
| Trasplante alogénico de células madre hematopoyéticas |
| Puede utilizarse en pacientes con enfermedad grave (como β-talasemia major) con donante compatible |
| Importante morbimortalidad si el donante no es un hermano compatible o se hace después de la infancia |
| Otras actuaciones terapéuticas |
| Dietéticas: suplementos de ácido fólico, evitar alimentos con excesivo hierro |
| Manejo de complicaciones reflejadas en la tabla 6 |
| Tratamientos en investigación |
| Terapia génica: posible alternativa al trasplante alogénico para β-talasemia TDT. Se trasfiere en la célula madre una extensión del gen de la β-globina por un vector lentiviral22 |
| Trampas de ligandos del receptor de activina IIA (luspatercept y sotatercept): promueven eritropoyesis |
AAS: ácido acetilsalicílico; HBPM: heparina de bajo peso molecular; TDT: talasemia dependiente de transfusiones; TNDT: talasemia no dependiente de transfusiones.
Fuente: elaboración propia.
La tabla 13 resume las principales opciones terapéuticas en otras anemias microcíticas.
Tratamiento de otras causas de anemia microcítica
| Anemia de enfermedad crónica14 |
| Tratar la enfermedad subyacente |
| Si existen déficits de hierro, ácido fólico o vitamina B12, tratarlos |
| Eritropoyetina y análogos: debe asociarse a suplementos de hierro (oral o intravenoso) para mantener niveles de ferritina ≥ 100 ng/mL e IST ≥ 20% |
| Transfusión: si el paciente está sintomático y no da tiempo a esperar al efecto de otros tratamientos o estos no funcionan |
| Anemia sideroblástica15 |
| Tratamiento sintomático de la anemia: transfusiones si son necesarias |
| Control de sobrecarga del hierro: monitorizarla y si aparece, realizar flebotomías (si anemia leve) o usar quelantes de hierro (si anemia severa) |
| Valorar si existe déficit de folatos y tratarlo |
| Valorar si existe déficit de cobre y tratarlo |
| Piridoxina: responden a ella dos tercios de AS ligadas a X y la AS por isoniacida |
| SMD con sideroblastos en anillo: se han utilizado eritropoyetina, G-CSF, lenalidomida, agentes hipometilantes, globulina antitimocítica, bloqueantes de la activación del TGFβ |
| Evitar esplenectomía |
| Trastornos genéticos del metabolismo del hierro4,5 |
| IRIDA: hierro intravenoso, algún caso responde a hierro oral más vitamina C |
| Mutaciones en SLC11A2: análogos de eritropoyetina |
| α-trasferrinemia: infusiones de plasma, vigilar y tratar sobrecarga de hierro |
| α-ceruloplasminemia: la anemia es leve y no necesita tratamiento. Puede haber sobrecarga tisular de hierro: valorar quelantes |
AS: anemia sideroblástica; CSF-G: factor estimulante de colonias de granulocitos; IRIDA: anemia ferropénica refractaria a hierro; IST: índice de saturación de transferrina; SMD: síndrome mielodisplásico; TGFβ: factor de crecimiento transformante β.
Fuente: elaboración propia.
